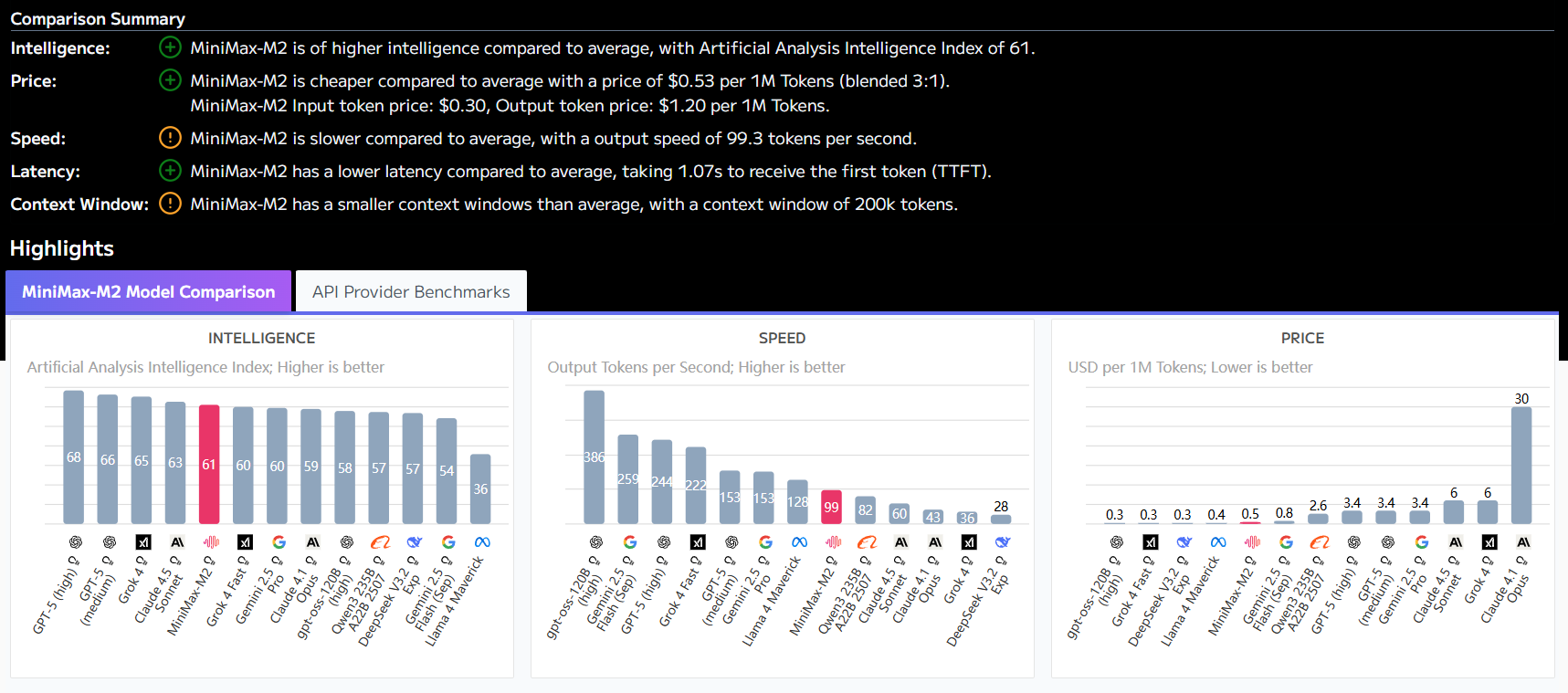
Evo 2's zero-sample variant effect prediction feature requires no additional training to assess gene variant effects, and the BRCA1 gene prediction process is as follows:
Prediction Steps:
- Prepare data: Storing reference and variant sequences in a list
ref_seqs = ["ATCG..."] # 参考序列
var_seqs = ["ATGG..."] # 变体序列 - Operational forecasts::
ref_scores = model.score_sequences(ref_seqs)
var_scores = model.score_sequences(var_seqs)
print(f"Reference score: {ref_scores}, Variant score: {var_scores}")
Results Interpretation:
The output contains the likelihood values of the reference and variant sequences. By comparing the differences between the two:
- Lower scores indicate that the variant may be harmful
- Slight changes indicate neutral variants
- Increased scores may be a gain-of-function variant
This function is particularly applicable to the prediction of pathogenicity of cancer-related genes (e.g. BRCA1), which provides a reference basis for precision medicine.
This answer comes from the articleEvo2: An Open Source BioAI Tool to Support Genome Modeling and DesignThe
May not be reproduced without permission:AI productivity tools " How does Evo 2 enable zero-sample variant effect prediction? BRCA1 is used as an example to illustrate